开发高效可靠的方法来对活细胞的基因组进行精确、有针对性的改变是生物医学研究人员的长期目标。近年来,一种基于来自化脓性链球菌的细菌CRISPR相关蛋白-9核酸酶(Cas9)的新工具引起了研究人员的极大兴趣[1]。这是多年来多次尝试操纵基因功能之后,包括同源重组[2]和RNA干扰(RNAi)[3]。特别是RNAi成为实验室的主要产品,能够廉价和高通量地研究基因功能[4,5],但由于仅提供基因功能的暂时抑制和不可预测的脱靶效应而受到阻碍[6]。最近靶向基因组修饰的其他方法-锌指核酸酶[ZFNs,[7]]和转录激活剂如效应核酸酶[TALENs[8]] - 使研究人员能够通过引入双链断裂来激活修复途径来产生永久性突变。这些方法的工程成本高昂且耗时,限制了它们的广泛使用,特别是对于大规模、高通量的研究。
什么是CRISPR/Cas9?
CRISPR(成簇的规则间隔的短回文重复序列)和CRISPR相关(Cas)基因的功能在选定细菌和古细菌的适应性免疫中至关重要,使生物体能够响应并消除入侵的遗传物质。这些重复序列最初是在1980年代在大肠杆菌中发现的[9],但直到2007年,Barrangou及其同事才证实它们的功能,他们证明嗜热链球菌可以通过将感染性病毒的基因组片段整合到其CRISPR基因座上而获得对噬菌体的抗性[10]。
三种类型的CRISPR机制已经被确认,其中II型是研究最多的。在这种情况下,来自病毒或质粒的入侵DNA被切割成小片段,并在一系列的短重复(约20 bps)中被纳入CRISPR基因座。该基因座被转录,然后转录物被处理以产生小RNA(crRNA - CRISPR RNA),这些小RNA被用来引导效应内切酶,根据序列互补性瞄准入侵的DNA(图1)[11]。
图1.体内Cas9:细菌适应性免疫
图1:[在获取阶段,外源DNA被整合到CRISPR位点的细菌基因组中。然后在crRNA生物发生过程中转录并加工成crRNA。在干扰过程中,Cas9核酸内切酶与crRNA复合,分离的tracrRNA切割含有与PAM序列相邻的20个核苷酸crRNA互补序列的外源DNA。]
一种Cas蛋白Cas9(也称为Csn1)已被证明是某些CRISPR机制(特别是II型CRISPR系统)的关键参与者。与其他CRISPR系统相比,II型CRISPR机制是独一无二的,因为基因沉默只需要一种Cas蛋白(Cas9)[12]。在II型系统中,Cas9参与crRNA的处理[12],并负责靶DNA的破坏[11]。Cas9在这两个步骤中的功能都依赖于两个核酸酶结构域的存在,一个位于氨基末端的RuvC样核酸酶结构域和一个位于蛋白质中部区域的HNH样核酸酶结构域[13]。为了实现位点特异性DNA识别和切割,Cas9必须与crRNA和单独的反式激活crRNA(tracrRNA或trRNA)复合,后者与crRNA部分互补[11]。tracrRNA是从编码多个前crRNA的初级转录本中crRNA成熟所必需的。这发生在存在RNase III和Cas9的情况下[12]。在靶DNA的破坏过程中,HNH和RuvC样核酸酶结构域切割两条DNA链,在相关crRNA转录本[11,14]中由20个核苷酸靶序列定义的位点产生双链断裂(DSB)。HNH结构域切割互补链,而RuvC结构域切割非互补链。Cas9的双链核酸内切酶活性还要求一个称为原间隔相关基序(PAM)的短保守序列(2-5 nts)紧跟在crRNA互补序列的3'-之后[15]。事实上,在没有PAM序列的情况下,即使是完全互补的序列也被Cas9-RNA忽略了[16]。
Cas9和CRISPR作为分子生物学的新工具
II型CRISPR核酸酶的简单性,只有三种必需的组分(Cas9以及crRNA和trRNA),使该系统适合基因组编辑。这种潜力在2012年由Doudna和Charpentier实验室实现[11]。基于前面描述的II型CRISPR系统,作者通过将trRNA和crRNA组合成单个合成单向导RNA(sgRNA)来开发一个简化的双组分系统。sgRNA编程的Cas9在指导靶向基因改变方面与使用单独的trRNA和crRNA编程的Cas9一样有效(图2-A)。
迄今为止,Cas9核酸酶的三种不同变体已被采用在基因组编辑方案中。第一种是野生型Cas9,它可以定点特异性切割双链DNA,导致双链断裂(DSB)修复机制的激活。DSB可以通过细胞非同源末端连接(NHEJ)途径修复[17],导致插入和/或缺失(插入缺失),从而破坏目标位点。或者,如果提供与目标位点同源的供体模板,则可以通过同源定向修复(HDR)途径修复DSB,从而允许进行精确的替换突变(图2-A)[17,18]。
Cong及其同事[1]通过开发一种仅具有切口酶活性的突变形式,称为Cas9D10A,使Cas9系统进一步提高了精度。这意味着它只切割一条DNA链,并且不会激活NHEJ。相反,当提供同源修复模板时,DNA修复仅通过高保真HDR途径进行,导致减少插入缺失突变[1,11,19]。当位点被设计用于产生相邻DNA切口的成对Cas9复合物靶向时,Cas9D10A在靶标特异性方面更具吸引力[20](参见图2-B中有关“配对切口酶”的更多详细信息)。
第三种变体是核酸酶缺陷的Cas9(dCas9,图2-C)[21]。突变H840A在HNH结构域和D10A在RuvC结构域灭活切割活性,但不阻止DNA结合[11,21]。因此,该变体可用于序列特异性靶向基因组的任何区域,而无需切割。相反,通过与各种效应结构域融合,dCas9可以用作基因沉默或激活工具[21,23-26]。此外,它可以用作可视化工具。例如,Chen及其同事使用与增强型绿色荧光蛋白(EGFP)融合的dCas9来可视化具有单个sgRNA的重复DNA序列或使用多个sgRNA的非重复位点[27]。
图2.CRISPR/Cas9系统应用
[图2-A:野生型Cas9核酸酶位点特异性切割双链DNA激活双链断裂修复机制。在没有同源修复模板的情况下,非同源末端连接会导致插入缺失破坏靶序列。或者,可以通过提供同源修复模板并利用同源定向修复途径来进行精确的突变和敲入。
图2-B:突变的Cas9产生位点特异性单链缺口。两个sgRNA可用于引入交错的双链断裂,然后可以进行同源定向修复。
图2-C:缺乏核酸酶的Cas9可以与各种效应结构域融合,从而实现特定的定位。例如,转录激活剂、阻遏因子和荧光蛋白。]
靶向效率和脱靶突变
靶向效率,或实现预期突变的百分比,是评估基因组编辑工具的最重要参数之一。Cas9的靶向效率与更成熟的方法,如TALENs或ZFNs[8]相比要好。例如,在人类细胞中,定制设计的ZFNs和TALENs只能达到1%到50%的效率[29-31]。相反,据报道,Cas9系统在斑马鱼[32]和植物中[33]的效率高达70%以上,在诱导多能干细胞中[34]的效率为2-5%。此外,Zhou及其同事能够在单细胞小鼠胚胎中把基因组靶向性提高到78%,并通过使用双sgRNAs同时靶向单个基因实现有效的种系传递[35]。
一种广泛使用的识别突变的方法是T7内切酶I突变检测法[36,37](图3)。这种检测方法检测异质双链DNA,它是包括所需突变的DNA链与野生型DNA链退火的结果[37]。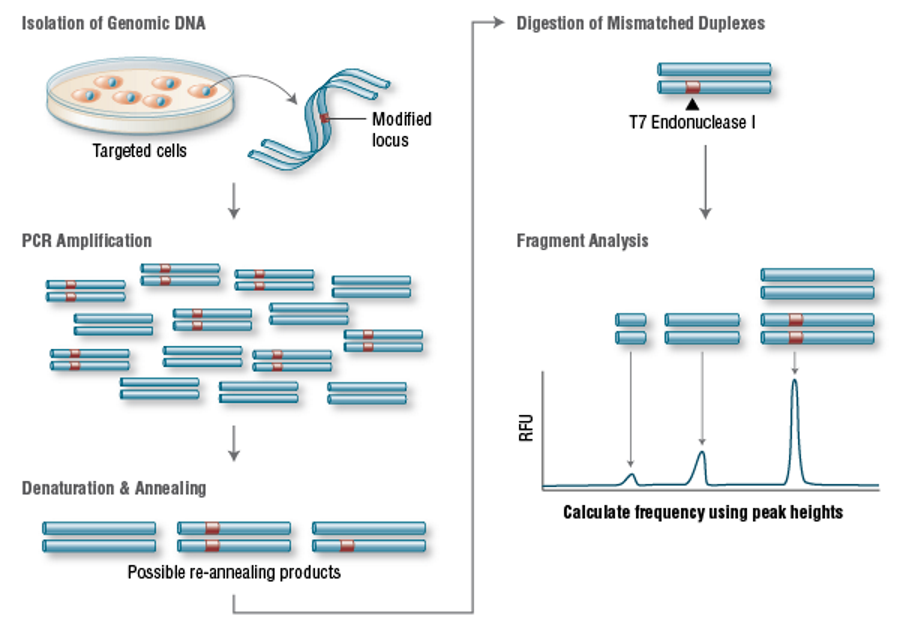
图3.T7核酸内切酶I靶向效率测定
[图3:基因组DNA用包围修饰位点的引物扩增。然后将PCR产物变性并重新退火,产生3种可能的结构。含有错配的双链体由T7核酸内切酶I消化。然后对DNA进行电泳分离,并使用片段分析来计算靶向效率。]
另一个重要参数是脱靶突变的发生率。这种突变很可能出现在与原始序列相比只有几个核苷酸差异的位点中,只要它们与PAM序列相邻。这是因为Cas9可以容忍原间隔区[36]内多达5个碱基错配或PAM序列中的单个碱基差异[38]。脱靶突变通常更难检测,需要全基因组测序才能完全排除它们。
通过使用截短的gRNA(在crRNA衍生序列内截短)或通过在5'末端添加两个额外的鸟嘌呤(G)核苷酸[28,37],最近对CRISPR系统进行了改进,以减少脱靶突变。研究人员试图最小化脱靶效应的另一种方法是使用“配对切口”[20]。该策略使用 D10A Cas9 和两个与靶位点相对链上的相邻区域互补的sgRNA(图2-B)。虽然这会在靶DNA中诱导DSB,但预计它只会在脱靶位置产生单个切口,因此导致最小的脱靶突变。通过利用计算来减少脱靶突变,几个小组开发了基于网络的工具,以促进识别潜在的CRISPR靶位点并评估其脱靶切割的潜力。例子包括CRISPR设计工具[38]和ZiFiT Targeter,版本4.2[39,40]。
作为基因组编辑和基因组靶向工具的应用
在2012年首次展示后[9],CRISPR/Cas9系统已被广泛采用。目前已经成功地用于许多细胞系和生物体中的重要基因,包括人类[34]、细菌[41]、斑马鱼[32]、秀丽隐杆线虫[42]、植物[34]、热带鱼[43]、酵母[44]、果蝇[45]、猴子[46]、兔子[47]、猪[42]、大鼠[48]和小鼠[49]。现在有几个小组已经利用这种方法,通过单一的gRNA,在一个特定的目标基因中引入单点突变(缺失或插入)[14,21,29]。用一对gRNA引导的Cas9核酸酶代替,也可以诱导大的缺失或基因组重排,如倒位或易位[50]。最近一个令人振奋的研究进展是使用CRISPR/Cas9系统的dCas9版本来针对蛋白域进行转录调控[26,51,52]、表观遗传修饰[25]和特定基因组位点的显微可视化[27]。
CRISPR/Cas9系统只需要重新设计crRNA即可改变靶标特异性。这与其他基因组编辑工具形成鲜明对比,包括ZFNs和TALENs,后者需要重新设计蛋白质- DNA界面。此外,CRISPR / Cas9通过生成用于基因组筛选的大型gRNA文库[51,53],可以快速进行全基因组的基因功能检测。
CRISPR/Cas9的未来
将Cas9开发成一套用于细胞和分子生物学研究的工具取得了显著进展,这可能是由于该系统的简单性,高效性和多功能性。在目前可用于精准基因组工程的设计核酸酶系统中,CRISPR/Cas系统是迄今为止最容易使用的。很显然,Cas9的潜力超出了DNA切割的范围,它对基因组位点特异性蛋白质募集的有用性可能只会受到我们的想象力的限制。
参考文献
1. Cong L., et al. (2013) Science, 339, 819–823.
2. Capecchi, M.R. (2005) Nat. Rev. Genet. 6, 507–512.
3. Fire, A., et al. (1998) Nature, 391, 806–811.
4. Elbashir, S.Mm, et al. (2002) Methods, 26, 199–213.
5. Martinez, J., et al. (2003) Nucleic Acids Res. Suppl. 333.
6. Alic, N, et al. (2012) PLoS One, 7, e45367.
7. Miller, J., et al. (2005) Mol. Ther. 11, S35–S35.
8. Mussolino, C., et al. (2011) Nucleic Acids Res. 39, 9283–9293.
9. Ishino, Y., et al. (1987) J. Bacteriol. 169, 5429–5433.
10. Barrangou, R., et al. (2007). Science, 315, 1709–1712.
11. Jinek, M., et al. (2012) Science, 337, 816–821.
12. Deltcheva, E., et al. (2011) Nature, 471, 602–607.
13. Sapranauskas, R., et al. (2011) Nucleic Acids Res. 39, 9275–9282.
14. Nishimasu, H., et al. (2014) Cell, doi:10.1016/j.cell.2014.02.001
15. Swarts, D.C., et al. (2012) PLoS One, 7:e35888.
16. Sternberg, S.H., et al. (2014) Nature, doi:10.1038/nature13011.
17. Overballe-Petersen, S., et al. (2013) Proc. Natl. Acad. Sci. U.S.A. 110,19860–19865.
18. Gong, C., et al. (2005) Nat. Struct. Mol. Biol. 12, 304–312.
19. Davis, L., Maizels, N. (2014) Proc. Natl. Acad. Sci. U S A, 111, E924–932.
20. Ran, F.A., et al. (2013) Cell, 154, 1380–1389.
21. Qi, L.S., et al. (2013) Cell, 152, 1173–1183.
22. Gasiunas, G., et al. (2012) Proc. Natl. Acad. Sci. U S A, 109, E2579–2586.
23. Maede, M.L., et al. (2013) Nat. Methods, 10, 977–979.
24. Gilbert, L.A., et al. (2013) Cell, 154, 442–451.
25. Hu, J., et al. (2014) Nucleic Acids Res. doi:10.1093/nar/gku109.
26. Perez-Pinera, P., et al. (2013) Nat. Methods, 10, 239–242.
27. Chen, B., et al. (2013) Cell, 155, 1479–1491.
28. Seung, W., et al. (2014) Genome Res. 24, 132–141.
29. Miller, J.C., et al. (2011). Nat. Biotechnol. 29, 143–148.
30. Mussolino, C., et al. (2011). Nucleic Acids Res. 39, 9283–9293.
31. Maeder, M.L., et al. (2008) Mol. Cell, 31, 294–301.
32. Hwang, W.Y., et al. (2013) PLoS One, 8:e68708.
33. Feng, Z., et al. (2013) Cell Res. 23, 1229–1232.
34. Mali, P., et al. (2013) Science, 339, 823–826.
35. Zhou, J., et al. (2014) FEBS J. doi:10.1111/febs.12735.
36. Fu, Y., et al. (2013) Nat. Biotechnol. 31, 822–826.
37. Fu, Y., et al. (2014) Nat Biotechnol. doi: 10.1038/nbt.2808.
38. Hsu, P.D., et al. (2013) Nat. Biotechnol. 31, 827–832.
39. Sander, J.D., et al. (2007) Nucleic Acids Res. 35, W599-605.
40. Sander, J.D., et al (2010) Nucleic Acids Res. 38, W462–468.
41. Pyne M.E., et al. (2015) Appl Environ Microbiol 81:5103–5114.
42. Oh J.H., et al. (2014) Nucleic Acids Res 42:e131.
43. Jiang W., et al. (2013) Nat Biotechnol 31:233–239.
44. Hai, T., et al. (2014) Cell Res. doi: 10.1038/cr.2014.11.
45. Guo, X., et al. (2014) Development, 141, 707–714.
46. DiCarlo, J.E., et al. (2013) Nucleic Acids Res. 41, 4336–4343.
47. Gratz, S.J., et al. (2014) Genetics, doi:10.1534/genetics.113.160713.
48. Niu, Y., et al. (2014) Cell, 156, 836–843.
49. Yang, D., et al. (2014) J. Mol. Cell Biol. 6, 97-99.
50. Ma, Y., et al. (2014) Cell Res. 24, 122–125.
51. Mashiko, D., et al. (2014) Dev. Growth Differ. 56, 122–129.
52. Gratz, S.J., et al. (2013) Fly, 249.
53. Mali, P., et al. (2013) Nat. Biotechnol. 31, 833–838.
 鲁公网安备 37021002001147号
鲁公网安备 37021002001147号








